Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
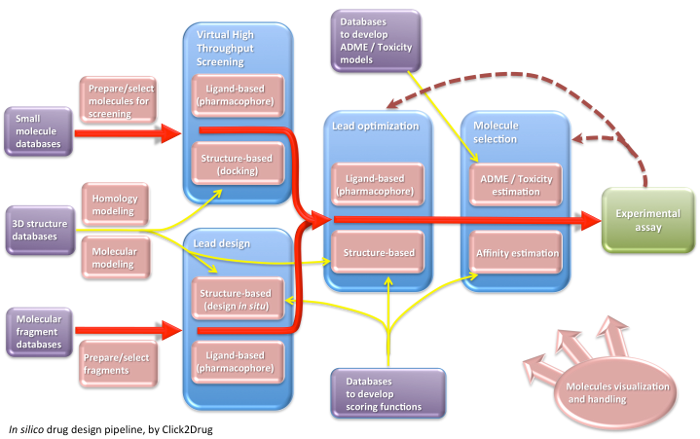
Show all links Hide all links
Binding free energy estimation
Software
- Hyde. Entirely new approach to assess binding affinities and contributions to binding of a complex, with a visual feedback at atomic detail. Hyde shows which regions of a complex contribute favorably and infavorably to the binding. Allows modifying a molecule interactively to optimize a complex and trigger new lead optimization ideas. Hyde is entirely based on physics-principles and has not been trained or calibrated on experimental data. Distributed by BioSolveIT.
- X-score. Program for computing the binding affinities of the given ligand molecules to their target protein. X-Score is released to the public for free.
- NNScore. Python script for computing binding free energies from PDBQT files of the receptor and the ligand, using a neural network approach. Free and open source. Developed by the McCammon Lab, UCSD.
Web services
- DSXONLINE. (Formerly DrugScoreONLINE). Web-based user interface for the knowledge-based scoring functions DSX.
- BAPPL server. Binding Affinity Prediction of Protein-Ligand (BAPPL) server computes the binding free energy of a non-metallo protein-ligand complex using an all atom energy based empirical scoring function.
- BAPPL-Z server. Binding Affinity Prediction of Protein-Ligand complex containing Zinc [ BAPPL-Z ] server computes the binding free energy of a zinc containing metalloprotein-ligand complex using an all atom energy based empirical scoring function.
- PreDDICTA. Predict DNA-Drug Interaction strength by Computing ΔTm and Affinity of binding.
- PharmaGist. Freely available web server for pharmacophore detection. The download version includes virtual screening capability.
- IC50-to-Ki converter. Computes Ki values from experimentally determined IC50 values for inhibitors of enzymes that obey classic Michaelis-Menten kinetics and of protein-ligand interactions
Databases
- CLiBE. Database containing information about Computed Ligand-Receptor Interaction Energy and other attributes such as energy components; ligand classification, functions and properties. Ligand structure is also included. Provided by the BioInformatics and Drug Design group of the National University of Singapore.
QSAR
Software
- cQSAR. A regression program that has dual databases of over 21,000 QSAR models. Distributed by BioByte.
- SeeSAR. Program for interactive, visual compound promotion and optimization. It include PD and PK parameters and can be linked to other modules for physicochemical and ADME. Distributed by Bio
- clogP. Program for calculating log Poct/water from structure. Distributed by BioByte.
- ClogP/CMR. Estimates Molar Refractivity and logP. Distributed by Tripos.
- Topomer CoMFA. 3D QSAR tool that automates the creation of models for predicting the biological activity or properties of compounds. Distributed by Tripos.
- QSARPro. QSAR software for evaluation of several molecular descriptors along with facility to build the QSAR equation (linear or non-linear regression) and use it for predicting the activities of test/new set of molecules. Performs 2D and 3D QSAR, and provides GQSAR, a group based QSAR approach establishing a correlation of chemical group variation at different molecular sites of interest with the biological activity. Works on LInux and Windows. Provided by VLife.
- MedChem Studio. (Formerly ClassPharmer). Cheminformatics platform supporting lead identification and prioritization, de novo design, scaffold hopping and lead optimization. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
- Surflex-Sim. Performs the alignment of molecules by maximizing their three-dimensional similarity. Surflex-Sim uses a surface-based morphological similarity function while minimizing the overall molecular volume of the aligned structures. Distributed by Tripos.
- QSAR with CoMFA. Builds statistical and graphical models that relate the properties of molecules (including biological activity) to their structures. Several structural descriptors can be calculated, including EVA and the molecular fields of CoMSIA. Distributed by Tripos.
- Almond. 3D-QSAR approach using GRid-INdependent Descriptors (GRIND). Starting with a set of 3D structures, Almond employs GRID3 force field to generate Molecular Interaction Fields (MIFs). The information in the MIFs is transformed to generate information-rich descriptors independent of the location of the molecules within the grid. Distributed by Tripos.
- GALAHAD. GA-based program to develop pharmacophore hypotheses and structural alignments from a set of molecules that bind at a common site. No prior knowledge of pharmacophore elements, constraints, or molecular alignment is required. Distributed by Tripos.
- Molegro Data Modeller. A program for building regression or classification models, performing feature selection and cross-validation, principal component analysis, high-dimensional visualization, clustering, and outlier detection. Provided by Qiagen.
- Hologram QSAR (HQSAR). Program using molecular holograms and PLS to generate fragment-based structure-activity relationships. Unlike other 3D-QSAR methods, HQSAR does not require alignment of molecules.
- cQSAR. A regression program that has dual databases of over 21,000 QSAR models. Distributed by BioByte.
- McQSAR. Free program to generates quantitative structure-activity relationships (QSAR equations) using the genetic function approximation paradigm. For Windows and Linux.
- CheS-Mapper. CheS-Mapper (Chemical Space Mapper) is a 3D-viewer for chemical datasets with small compounds. The tool can be used to analyze the relationship between the structure of chemical compounds, their physico-chemical properties, and biological or toxic effects. CheS-Mapper embedds a dataset into 3D space, such that compounds that have similar feature values are close to each other. It can compute a range of descriptors and supports clustering and 3D alignment. It is an open-source Java application, based on the Java libraries Jmol, CDK, WEKA, and utilizes OpenBabel and R. Developed and proposed by the Universität Mainz, Germany.
- Open3DQSAR. Program aimed at high-throughput generation and chemometric analysis of molecular interaction fields (MIFs). Free open source software. For Windows, Linux and Mac.
- PaDEL-Descriptor. Free software to calculate molecular (1875) descriptors and (12) fingerprints. Can be used from command lines or GUI. Developed by the National University of Singapore.
- Codessa. Derives descriptors using quantum mechanical results from AMPAC. These descriptors are then used to develop QSAR/QSPR models.
- CDK Descriptor Calculator GUI. Free and open source GUI to CDK to calculate molecular descriptors.
- BlueDesc. Free and open source molecular descriptor calculator. Converts an MDL SD file into ARFF and LIBSVM format for machine learning and data mining purposes using CDK and JOELib2. Provided by the Tuebingen University.
- KeyRecep. Estimates the characteristics of the binding site of the target protein by superposing multiple active compounds in 3D space so that the physicochemical properties of the compounds match maximally with each other. Can be used to estimate activities and vHTS. Distributed by IMMD.
- OpenMolGRID. Uses a Grid approach to deal with large-scale molecular design and engineering problems. The methodology used relies on Quantitative Structure Property/Activity Relationships (QSPR/QSAR).
- Molconn-Z. Standard program for generation of Molecular Connectivity, Shape, and Information Indices for Quantitative Structure Activity Relationship (QSAR) Analyses.
- CODESSA Pro. Program for developing quantitative structure-activity/property relationships (QSAR/QSPR. Distributed by CompuDrug.
- MCASE. Machine learning approach to automatically evaluate compounds/activity data set and identify the structural features responsible for activity (biophores). It then creates organized dictionaries of these biophores and develops ad hoc local QSAR correlations. Distributed by MultiCASE.
- hint!. (Hydropathic INTeractions). Estimates LogP for modeled molecules or data files, numerically and graphically evaluates binding of drugs or inhibitors into protein structures and scores DOCK orientations, constructs hydropathic (LOCK and KEY) complementarity maps that can be used to predict a substrate from a known receptor or protein structure or to propose the hydropathic structure from known agonists or antagonists, and evaluates/predicts effects of site-directed mutagenesis on protein structure and stability.
- smirep. System for predicting the structural activity of chemical compounds.
Databases
- MOLE db. Molecular Descriptors Data Base is a free on-line database comprised of 1124 molecular descriptors calculated for hundreds of thousands of molecules.
- ChemDB/Datasets. Experimentally annotated subsets of the ChemDB for machine learning and searching experiments.
- Datasets from the Milano Chemometrics and QSAR Research Group. References Data Sets
- OCHEM Database. Online database of experimental measurements integrated with a modeling environment. User can submit experimental data or use the data uploaded by other users to build predictive QSAR models for physical-chemical or biological properties. Provided by eADMET GmbH and the Institute of Bioinformatics & Systems Biology at Helmholtz Zentrum München.
- ChemSAR. Free web-based pipelining platform for classification models of small molecules. It includes validation and standardization of chemical structures, calculation of descriptots (1D, 2D and FP), feature selection, model building and interpretation. Developed by the School of Pharmaceutical Sciences, Central South University, China.
- Chembench. Free portal that enables researchers to mine available chemical and biological data. It includes robust model builders, property and activity predictors, virtual libraries of available chemicals with predicted biological and drug-like properties, and special tools for chemical library design. Provided with registration by the Carolina Exploratory Center for Cheminformatics Research, USA.
Web services
- OCHEM. (Online Chemical Modeling Environment project). Online database of experimental measurements integrated with a modeling environment. User can submit experimental data or use the data uploaded by other users to build predictive QSAR models for physical-chemical or biological properties. Provided by eADMET GmbH and the Institute of Bioinformatics & Systems Biology at Helmholtz Zentrum München.
- E-Dragon. Online version of DRAGON, which is an application for the calculation of molecular descriptors developed by the Milano Chemometrics and QSAR Research Group. These descriptors can be used to evaluate molecular structure-activity or structure-property relationships, as well as for similarity analysis and highthroughput screening of molecule databases. Provided by the Virtual Computational Chemistry Laboratory.
- Pattern Match Counter. Counts Functional Groups (sub-structures) in molecules.
- Pattern Count Screen. Screens by Functional Groups.
- Partial Least Squares Regression (PLSR). Generates model construction and prediction of activity/property using the Partial Least Squares (PLS) regression technique. Provided by the Virtual Computational Chemistry Laboratory.
- XScore-LogP. Calculates the octanol/water partition coefficient for a drug, based on a feature of the X-Score program.
- 3-D QSAR. 3-D QSAR MODELS DATABASE for Virtual Screening. users can process their own molecules by drawing or uploading them to the server and selecting the target for the virtual screening and biological activity prediction.
- MOLFEAT. Web service to compute molecular fingerprints and molecular descriptors of molecules from their 3D structures, and for computing activity of compounds of specific chemical types against selected targets based on published Quantitative Structure-Activity Relationship (QSAR) models. Currently covers 1,114 fingerprints, 3,977 molecular descriptors, and 23 QSAR models for 16 chemical types against 14 targets. Maintained by the University of Singapore.