Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
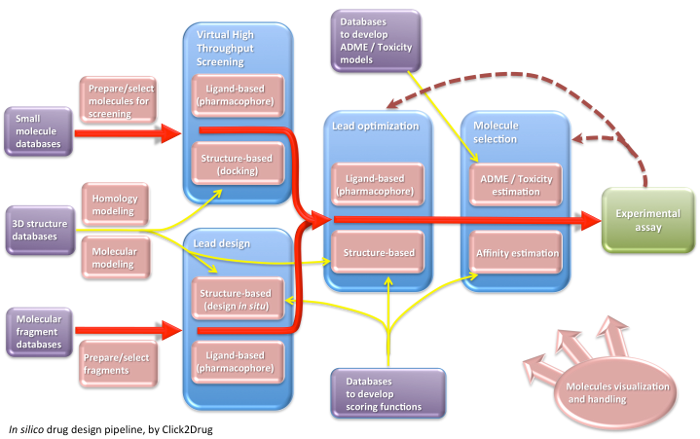
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
Show all links Hide all links
Chemical structure representations
3D viewers
- VEGA ZZ. Visualization application and molecular modeling toolkit (Molecular mechanics and dynamics, structure-based screening). Free for non-profit academic uses. Provided by the Drug Design Laboratory of the University of Milano.
Binding site prediction
Software
- MED-SuMo. Program for macromolecules surface similarity detection. Searches into 3D databases, find similar binding surfaces and generate 3D superpositions based on common surface chemical features and similar shape. Can be used for site mining, drug repurposing and site classification at PDB scale. Distributed by MEDIT.
- TRAPP. TRAnsient Pockets in Proteins (TRAPP) is a web server for the analysis of transient binding pockets in proteins. Contrarily to many tools, it is not intended for ligand binding pocket identification per se, but rather to predict significant changes in the spatial and physicochemical properties of a given pocket that may arise due to the protein's flexibility (both backbone and side chains). Several capabilities of visualization and analysis have been developed and are provided by the Molecular and Cellular Modeling group at Heidelberg Institute for Theoretical Studies, Germany.
- CAVER. Software tool for analysis and visualisation of tunnels and channels in protein structures. Provided by the Masaryk University.
- fpocket. Open source protein pocket (cavity) detection algorithm based on Voronoi tessellation. Developed in the C programming language and currently available as command line driven program. fpocket includes two other programs (dpocket & tpocket) that allow you to extract pocket descriptors and test own scoring functions respectively. Also contains a druggability prediction score.
- GHECOM. Program for finding multi-scale pockets on protein surfaces using mathematical morphology. Free open source.
- LIGSITEcsc. Program for the automatic identification of pockets on protein surface using the Connolly surface and the degree of conservation.
- SURFNET. Generates surfaces and void regions between surfaces from coordinate data supplied in a PDB file.
- SiteHound. Identifies ligand binding sites by computing interactions between a chemical probe and a protein structure. The input is a PDB file of a protein structure, the output is a list of “interaction energy clusters” corresponding to putative binding sites.
- ICM-PocketFinder. Binding site predictor based on calculating the drug-binding density field and contouring it at a certain level. Provided by Molsoft.
- SiteMap. Program for binding site identification. Distributed by Schrodinger.
- MSPocket. Orientation independent program for the detection and graphical analysis of protein surface pockets. A MSPocket plugin for PyMOL provides a graphical user interface for runing MSPocket and render its results in PyMOL. It is included in the download. Free and open source.
- POCASA. (POcket-CAvity Search Application). Automatic web service that implements the algorithm named Roll which can predict binding sites by detecting pockets and cavities of proteins of known 3D structure. Maintained by the Hokkaido University.
- Phosfinder. Method for the prediction of phosphate-binding sites in protein structures. provided by the University of Rome.
- VOIDOO. Software to find cavities and analyse volumes.
- FunFOLDQA. Program to assess the quality ligand binding site residue predictions based on 3D models of proteins. Free program written in java. Developped by the University of Reading.
- LISE. Free and open source program for ligand Binding Site Prediction Using Ligand Interacting and Binding Site-Enriched Protein Triangles. Exists as a web service. Provided by the Institute of Biomedical Sciences, Academia Sinica.
- PDBinder. Free program for the identification of small ligand-binding sites in a protein structure. webPDBinder searches a protein structure against a library of known binding sites and a collection of control non-binding pockets. Exists as a web service. Provided by the University of Roma 2, Italy.
- eFindSite. Ligand binding site prediction and virtual screening algorithm that detects common ligand binding sites in a set of evolutionarily related proteins identified by 10 threading/fold recognition methods. Exists as a web service. Provided by the Louisiana State University, Computational Systems Biology Group.
- POVME. Free and open source program for measuring binding-pocket volumes. Developed by the National Biomedical Computation Resource.
- SiteEngine. Program to predict regions that can potentially function as binding sites. The methods is based on recognition of geometrical and physico-chemical environments that are similar to known binding sites. Exists as a web service. Provided by the structural Bioinformatics group at Tel-Aviv University.
- SVILP_ligand. General method for discovering the features of binding pockets that confer specificity for particular ligands. Provided by the Computational Bioinformatics Laboratory, Imperial College London.
Databases
- sc-PDB. Annotated Database of Druggable Binding Sites from the Protein DataBank. Provided by the university of Strasbourg.
- CASTp. Computed Atlas of Surface Topography of proteins. Provides identification and measurements of surface accessible pockets as well as interior inaccessible cavities, for proteins and other molecules. castP server uses the weighted Delaunay triangulation and the alpha complex for shape measurements.
- Pocketome. Encyclopedia of conformational ensembles of all druggable binding sites that can be identified experimentally from co-crystal structures in the Protein Data Bank.
- PDBe motifs and Sites. Can be used to examine the characteristics of the binding sites of single proteins or classes of proteins such as Kinases and the conserved structural features of their immediate environments either within the same specie or across different species.
- LigASite. Dataset of biologically relevant binding sites in protein structures. It consists of proteins with one unbound structure and at least one structure of the protein-ligand complex. Both a redundant and a non-redundant (sequence identity lower than 25%) version is available.
- PROtein SURFace ExploreR. Contains information about structural similarities with respect to the query surfaces. A pocket search algorithm detected 48,347 potential ligand binding sites from the 9,708 non-redundant protein entries in the PDB database. All-against-all structural comparison was performed for the predicted sites, and the similar sites with the Z-score ≥ 2.5 were selected. These results can be accessed by the PDB code or ligand name.
- fPOP. Footprinting protein functional surfaces by comparative spatial patterns. Database of the protein functional surfaces identified by shape analysis.
- PDBSITE. Database on protein active sites and their spatial environment. Provided by GeneNetworks.
- LigBase. Database of ligand binding proteins aligned to structural templates. The structural templates are taken from the PDB, 3D models of the aligned sequences are provided ModBase, and pairwise sequence alignments are provided by CE. Multiple Structural Alignments are built on the fly within LigBase from a series of pairwise alignments. Ligand diagrams are generated with the program Ligplot. Maintained by Andrej Sali at the University of California, San Francisco.
Web services
- 3DLigandSite. Automated method for the prediction of ligand binding sites. Provided by the Imperial London College.
- metaPocket. Meta server to identify pockets on protein surface to predict ligand-binding sites.
- PockDrug. A methodology tehat predicts pocket druggability, efficient on both; estimated pockets guided by the ligand proximity (extracted by proximity to a ligand from a holo protein structure using several thresholds) and estimated pockets not guided by the ligand proximity (based on amino atoms that form the surface of potential binding cavities).. Developed and maintained by the University Paris-Diderot, France.
- PocketQuery. Protein-protein interaction (PPI) inhibitor starting points from PPI structure. Quickly identify a small set of residues at a protein interface that are suitable starting points for small-molecule design. Provided by the University of Pittsburgh.
- PASS. Program for tentative identification of drug interaction pockets from protein structure.
- DEPTH. Web server to compute depth and predict small-molecule binding cavities in proteins
- fpocket web server. Open source protein pocket (cavity) detection algorithm based on Voronoi tessellation. Developed in the C programming language and currently available as command line driven program. fpocket includes two other programs (dpocket & tpocket) that allow you to extract pocket descriptors and test own scoring functions respectively. Also contains a druggability prediction score.
- Nucleos. Nucleos is a webserver for the identification of nucleotide-binding sites based on the concept of nucleotide modularity. Nucleos identifies binding sites for nucleotide modules (namely the nucleobase, the carbohydrate and the phosphate) and then combines them in order to build the complete binding sites for different types of nucleotides (e.g. ADP or FAD). Provided by the University of Roma 2, Italy.
- wwwPDBinder. Web server for the identification of small ligand-binding sites in a protein structure. webPDBinder searches a protein structure against a library of known binding sites and a collection of control non-binding pockets. Exists as a standalone program. Provided by the University of Roma 2, Italy.
- IsoMIF. IsoMIF identifies binding site molecular interaction field similarities between proteins. The IsoMIF Finder Interface allows you to identify binding site molecular interaction field (MIF) similarities between a query structure and a database of pre-calculated MIFs or you own custom PDB entries. Developed by the University of Sherbrooke, Canada.
- LISE. Ligand Binding Site Prediction Using Ligand Interacting and Binding Site-Enriched Protein Triangles. Exists as a standalone program. Provided by the Institute of Biomedical Sciences, Academia Sinica.
- eFindSite. Web server for ligand binding site prediction and virtual screening algorithm that detects common ligand binding sites in a set of evolutionarily related proteins identified by 10 threading/fold recognition methods. Exists as standalone program. Provided by the Louisiana State University, Computational Systems Biology Group.
- Active Site Prediction. Web server for computing the cavities in a given protein. Provided by the Supercomputing Facility for Bioinformatics & Computational Biology, IIT Delhi.
- GHECOM web server. Web server for finding multi-scale pockets on protein surfaces using mathematical morphology.
- LIGSITEcsc web server. Web server for the automatic identification of pockets on protein surface using the Connolly surface and the degree of conservation.
- ProBis. Web server for detection of structurally similar binding sites. Maintained by the National Institute of Chemistry, Ljubljana, Slovenia.
- ProBiS-CHARMMing. Web server for detection of structurally similar binding sites, plus minimization of predicted protein-ligand complexes and their interaction energy calculation. Maintained by the National Institute of Health, USA.
- FunFOLD. Web server to predict likely ligand binding site residues for a submitted amino acid sequence.
- CAVER. Software tool for analysis and visualisation of tunnels and channels in protein structures. Provided by the Masaryk University.
- SuMo. Screens the Protein Data Bank (PDB) for finding ligand binding sites matching your protein structure or inversely, for finding protein structures matching a given site in your protein. Provided freely by the Pole Bioinformatique Lyonnais.
- IBIS. (Inferred Biomolecular Interactions Server). For a given protein sequence or structure query, IBIS reports physical interactions observed in experimentally-determined structures for this protein. IBIS also infers/predicts interacting partners and binding sites by homology, by inspecting the protein complexes formed by close homologs of a given query.
- PocketDepth. Depth based algortihm for identification of ligand binding sites.
- Screen2. Tool for identifying protein cavities and computing cavity attributes that can be applied for classification and analysis.
- SiteHound-web. Identifies ligand binding sites by computing interactions between a chemical probe and a protein structure. The input is a PDB file of a protein structure, the output is a list of “interaction energy clusters” corresponding to putative binding sites. Maintained by the Sanchez lab, at the Mount Sinai School of Medicine, NY, USA.
- SiteComp. Web server providing three major types of analysis based on molecular interaction fields: binding site comparison, binding site decomposition and multi-probe characterization. Maintained by the Sanchez lab, at the Mount Sinai School of Medicine, NY, USA.
- ConCavity. Ligand binding site prediction from protein sequence and structure.
- SplitPocket. Prediction of binding sites for unbound structures.
- PepSite 2. Web service for the prediction of peptide binding sites on protein surfaces. Developed and maintained by the Russel Lab, University of Heidelberg.
- MolAxis. Web server for the identification of channels in macromolecules.
- PDBSiteScan. Tool for search for functional sites in protein tertiary structures. Developed in collaboration with Institute of Cytology and Genetics, Novosibirsk.
- MultiBind. (Multiple Alignment of Protein Binding Sites). Prediction tool for protein binding sites. Users input a set of protein-small molecule complexes and MultiBind predicts the common physio-chemical patterns responsible for protein binding. Exists as a standalone program. Provided by the structural Bioinformatics group at Tel-Aviv University.
- SiteEngine. Web service to predict regions that can potentially function as binding sites. The methods is based on recognition of geometrical and physico-chemical environments that are similar to known binding sites. Exists as a standalone program. Provided by the structural Bioinformatics group at Tel-Aviv University.
Docking
Software
- Autodock. Free open source EA based docking software. Flexible ligand. Flexible protein side chains. Maintained by the Molecular Graphics Laboratory, The Scripps Research Institute, la Jolla.
- DOCK. Anchor-and-Grow based docking program. Free for academic usage. Flexible ligand. Flexible protein. Maintained by the Soichet group at the UCSF.
- GOLD. GA based docking program. Flexible ligand. Partial flexibility for protein. Product from a collaboration between the university of Sheffield, GlaxoSmithKline plc and CCDC.
- Glide. Exhaustive search based docking program. Exists in extra precision (XP), standard precision (SP) and virtual High Throughput Screening modes. Ligand and protein flexible. Provided by Schrödinger.
- Itzamna. Itzamna is a docking program, identifying active compounds for a given target. You can upload a protein and a docking is performed, either against an in-house database containing more than a million active compounds, or against a user-defined library. Provided by Mind The Byte.
- SCIGRESS. Desktop/server molecular modeling software suite employing linear scaling semiempirical quantum methods for protein optimization and ligand docking. Developed and distributed by Fujitsu, Ltd.
- GlamDock. Docking program based on a Monte-Carlo with minimization (basin hopping) search in a hybrid interaction matching / internal coordinate search space. Part of the Chil2 suite. Open for general research.
- FlexAID. A small-molecule docking algorithm that accounts for target side-chain flexibility and utilizes a soft scoring function. The pairwise energy parameters were derived from a large dataset of true positive poses and negative decoys from the PDBbind database through an iterative process using Monte Carlo simulations. Precompiled Linux, MacOS and Windows versions are made available by the University of Sherbrooke, Canada.
- GEMDOCK. Generic Evolutionary Method for molecular DOCKing. Program for computing a ligand conformation and orientation relative to the active site of target protein==== Docking - Software ====
- iGEMDOCK. Graphic environment for the docking, virtual screening, and post-screening analysis. Free for non commercial researches. For Windows and Linux.
- HomDock. Progam for similarity-based docking, based on a combination of the ligand based GMA molecular alignment tool and the docking tool GlamDock. Part of the Chil2 suite. Open for general research.
- ICM. Docking program based on pseudo-Brownian sampling and local minimization. Ligand and protein flexible. Provided by MolSoft.
- FlexX, Flex-Ensemble (FlexE). Incremental build based docking program. Flexible ligand. Protein flexibility through ensemble of protein structure. Provided by BioSolveIT.
- Fleksy. Program for flexible and induced fit docking using receptor ensemble (constructed using backbone-dependent rotamer library) to describe protein flexibility. Provided by the Centre for Molecular and Biomolecular Informatics, Radboud University Nijmegen.
- FITTED. (Flexibility Induced Through Targeted Evolutionary Description). Suite of programs to dock flexible ligands into flexible proteins. This software relies on a genetic algorithm to account for flexibility of the two molecules and location of water molecules, and on a novel application of a switching function to retain or displace water molecules and to form potential covalent bonds (covalent docking) with the protein side-chains. Part of the Molecular FORECASTER package and FITTED Suite. Free for an academic site license (excluding cluster).
- FORECASTER. Standalone interface that contains applications to perform docking and more. It includes the FITTED docking program, the sites of metabolism prediction IMPACTS, and the accessory programs to work with the proteins and the ligands. It comes with a java-based graphical interface that integrated all the program into workflows. Provided by Molecular Forecaster Inc.
- VLifeDock. Multiple approaches for protein - ligand docking. Provides three docking approches: Grid based docking, GA docking and VLife's own GRIP docking program. Several scoring functions can be used: PLP score, XCscore and Steric + Electrostatic score. Available for Linux and Windows. Provided by VLife.
- ParaDockS. (Parallel Docking Suite). Free, open source program, for docking small, drug-like molecules to a rigid receptor employing either the knowledge-based potential PMF04 or the empirical energy function p-Score.
- Molegro Virtual Docker. Protein-ligand docking program with support for displaceable waters, Induced-fit-docking, user-defined constraints, molecular alignment, ligands-based screening, and KNIME workflow integration. Distributed by Qiagen.
- DAIM-SEED-FFLD. Free open source fragment-based docking suite. The docking is realized in three steps. DAIM (Decomposition And Identification of Molecules) decomposes the molecules into molecular fragments that are docked using SEED (Program for docking libraries of fragments with solvation energy evaluation). Finally, the molecules are reconstructed ''in situ'' from the docked fragments using the FFLD program (Program for fragment-based flexible ligand docking). Developed and maintained by the Computational Structural Biology of ETH, Zurich, Switzerland.
- Autodock Vina. MC based docking software. Free for academic usage. Flexible ligand. Flexible protein side chains. Maintained by the Molecular Graphics Laboratory, The Scripps Research Institute, la Jolla.
- VinaMPI. Massively parallel Message Passing Interface (MPI) program based on the multithreaded virtual docking program AutodockVina. Free and open source. Provided by the University of Tennessee.
- FlipDock. GA based docking program using FlexTree data structures to represent a protein-ligand complex. Free for academic usage. Flexible ligand. Flexible protein. Developed by the Department of Molecular Biology at the Scripps Research Institute, la Jolla.
- PharmDock. A protein pharmacophore-based docking program. PharmDock and a PyMOL plugin are made freely available by the Purdue University, West Lafayette, USA.
- FRED. FRED performs a systematic, exhaustive, nonstochastic examination of all possible poses within the protein active site, filters for shape complementarity and pharmacophoric features before selecting and optimizing poses using the Chemgauss4 scoring function. Provided by OpenEye scientific software.
- POSIT. POSIT uses the information from bound ligands to improve pose prediction. Using a combination of approaches, including structure generation, shape alignment and flexible fitting, a ligand of interest is compared to bound ligands and its similarity to such both guides the nature of the applied algorithm and produces an estimate. Both 2D and 3D similarity measures are used in this reliability index. Provided by OpenEye scientific software.
- HYBRID. Docking program similar to FRED, except that it uses the Chemical Gaussian Overlay (CGO) ligand-based scoring function. Provided by OpenEye scientific software.
- idock. Free and open source multithreaded virtual screening tool for flexible ligand docking for computational drug discovery. Developed by the Chinese university of Hong Kong.
- POSIT. Ligand guided pose prediction. POSIT uses bound ligand information to improve pose prediction. Using a combination of several approaches, including structure generation, shape alignment and flexible fitting, it produces a predicted pose whose accuracy depends on similarity measures to known ligand poses. As such, it produces a reliability estimate for each predicted pose. In addition, if provided with a selection of receptors from a crystallographic series, POSIT will automatically determine which receptor is best suited for pose prediction. Provided by OpenEye scientific software.
- Rosetta Ligand. Monte Carlo minimization procedure in which the rigid body position and orientation of the small molecule and the protein side-chain conformations are optimized simultaneously. Free for academic and non-profit users.
- Surflex-Dock. Docking program based on an idealized active site ligand (a protomol), used as a target to generate putative poses of molecules or molecular fragments, which are scored using the Hammerhead scoring function. Distributed by Tripos.
- CDocker. CHARMm based docking program. Random ligand conformations are generated by molecular dynamics and the positions of the ligands are optimized in the binding site using rigid body rotations followed by simulated annealing. Provided by Accelrys.
- LigandFit. CHARMm based docking program. Ligand conformations generated using Monte-Carlo techniques are initially docked into an active site based on shape, followed by further CHARMm minimization. Provided by Accelrys.
- rDock. Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. Free and open source.Developed by the University of Barcelona.
- KIN. Kin is a blind-docking technology. All potential cavities of a given protein are predicted, and a query molecule is docked inside each of them, sorting results by scoring function. Distributed by Mind The Byte.
- Lead Finder. program for molecular docking, virtual screening and quantitative evaluation of ligand binding and biological activity.Distributed by Moltech. For Windows and linux.
- YASARA Structure. Adds support for small molecule docking to YASARA View/Model/Dynamics using Autodock and Fleksy. Provided by YASARA.
- ParaDockS. ParaDockS includes algorithms for protein-ligand docking and is organized that every newly developed scoring function can be immediately implemented. Furthermore, interaction-based classifier, trained on a target-specific knowledge base can be used in a post-docking filter step. An implementation and validation of target-biased scoring methods within the open-source docking framework is implemented. developed and provided free of charge by the University of Halle-Wittenberg, Germany.
- GalaxyDock. Protein-ligand docking program that allows flexibility of pre-selected side-chains of ligand. Developed by the Computational Biology Lab, Department of Chemistry, Seoul National University.
- MS-Dock. Free multiple conformation generator and rigid docking protocol for multi-step virtual ligand screening.
- BetaDock. Molecular docking simulation software based on the theory of Beta-complex.
- ADAM. Automated docking tool. Can be used for vHTS. Distributed by IMMD.
- hint!. (Hydropathic INTeractions). Estimates LogP for modeled molecules or data files, numerically and graphically evaluates binding of drugs or inhibitors into protein structures and scores DOCK orientations, constructs hydropathic (LOCK and KEY) complementarity maps that can be used to predict a substrate from a known receptor or protein structure or to propose the hydropathic structure from known agonists or antagonists, and evaluates/predicts effects of site-directed mutagenesis on protein structure and stability.
- DockVision. Docking package including Monte Carlo, Genetic Algorithm, and database screening docking algorithms.
- PLANTS. (Protein-Ligand ANT System). Docking algorithm based on a class of stochastic optimization algorithms called ant colony optimization (ACO). In the case of protein-ligand docking, an artificial ant colony is employed to find a minimum energy conformation of the ligand in the binding site. These ants are used to mimic the behavior of real ants and mark low energy ligand conformations with pheromone trails. The artificial pheromone trail information is modified in subsequent iterations to generate low energy conformations with a higher probability. Developed by the Konstanz university.
- EADock. Hybrid evolutionary docking algorithm with two fitness functions, in combination with a sophisticated management of the diversity. EADock is interfaced with the CHARMM package for energy calculations and coordinate handling.
- EUDOC. Program for identification of drug interaction sites in macromolecules and drug leads from chemical databases.
- FLOG. Rigid body docking program using databases of pregenerated conformations. Developed by the Merck Research Laboratories.
- Hammerhead. Automatic, fast fragment-based docking procedure for flexible ligands, with an empirically tuned scoring function and an automatic method for identifying and characterizing the binding site on a protein.
- ISE-Dock. Docking program which is based on the iterative stochastic elimination (ISE) algorithm.
- ASEDock. Docking program based on a shape similarity assessment between a concave portion (i.e., concavity) on a protein and the ligand. Developed by yoka Systems.
- HADDOCK. HADDOCK (High Ambiguity Driven biomolecular DOCKing) is an approach that makes use of biochemical and/or biophysical interaction data such as chemical shift perturbation data resulting from NMR titration experiments, mutagenesis data or bioinformatic predictions. First developed from protein-protein docking, it can also be applied to protein-ligand docking. Developed and maintained by the Bijvoet Center for Biomolecular Research, Netherlands.
- Computer-Aided Drug-Design Platform using PyMOL. PyMOL plugins providing a graphical user interface incorporating individual academic packages designed for protein preparation (AMBER package and Reduce), molecular mechanics applications (AMBER package), and docking and scoring (AutoDock Vina and SLIDE).
- Autodock Vina plugin for PyMOL. Allows defining binding sites and export to Autodock and VINA input files, doing receptor and ligand preparation automatically, starting docking runs with Autodock or VINA from within the plugin, viewing grid maps generated by autogrid in PyMOL, handling multiple ligands and set up virtual screenings, and set up docking runs with flexible sidechains.
- GriDock. Virtual screening front-end for AutoDock 4. GriDock was designed to perform the molecular dockings of a large number of ligands stored in a single database (SDF or Zip format) in the lowest possible time. It take the full advantage of all local and remote CPUs through the MPICH2 technology, balancing the computational load between processors/grid nodes. Provided by the Drug Design Laboratory of the University of Milano.
- DockoMatic. GUI application that is intended to ease and automate the creation and management of AutoDock jobs for high throughput screening of ligand/receptor interactions.
- BDT. Graphic front-end application which control the conditions of AutoGrid and AutoDock runs. Maintained by the Universitat Rovira i Virgili,
Web services
- SwissDock. SwissDock, a web service to predict the molecular interactions that may occur between a target protein and a small molecule.
- DockingServer. DockingServer offers a web-based, easy to use interface that handles all aspects of molecular docking from ligand and protein set-up.
- 1-Click Docking. Free online molecular docking solution. Solutions can be visualized online in 3D using the WebGL/Javascript based molecule viewer of GLmol. Provided by Mcule.
- Blaster. Public access service for structure-based ligand discovery. Uses DOCK as the docking program and various ZINC Database subsets as the database.Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- Docking At UTMB. Web-driven interface for performing structure-based virtual screening with AutoDock Vina. Maintained by the Watowich lab at the University of Texas Medical Branch.
- Blind Docking Server. A web-based tool to run molecular docking on the whole surface of the protein. The calculation are based on a customized version of Autodock Vina. Results and analyses can be explored on-line or downloaded. Some services are free, other are cost-based. Developed by the Bioinformatics and High Performance Computing Research group at the Universidad Católica San Antonio de Murcia (UCAM), Spain.
- Pardock. All-atom energy based Monte Carlo, rigid protein ligand docking, implemented in a fully automated, parallel processing mode which predicts the binding mode of the ligand in receptor target site. Maintained by the Supercomputing Facility for Bioinformatics & Computational Biology, IIT Delhi.
- FlexPepDock. High-resolution peptide docking (refinement) protocol, implemented within the Rosetta framework. The input for this server is a PDB file of a complex between a protein receptor and an estimated conformation for a peptide.
- PatchDock. Web server for structure prediction of protein-protein and protein-small molecule complexes based on shape complementarity principles.
- MEDock. Maximum-Entropy based docking web server for efficient prediction of ligand binding sites.
- BSP-SLIM. Web service for blind molecular docking method on low-resolution protein structures. The method first identifies putative ligand binding sites by structurally matching the target to the template holo-structures. The ligand-protein docking conformation is then constructed by local shape and chemical feature complementarities between ligand and the negative image of binding pockets. Provided by the University of Michigan.
- BioDrugScreen. Computational drug design and discovery resource and server. The portal contains the DOPIN (Docked Proteome Interaction Network) database constituted by millions of pre-docked and pre-scored complexes from thousands of targets from the human proteome and thousands of drug-like small molecules from the NCI diversity set and other sources. The portal is also a server that can be used to (i) customize scoring functions and apply them to rank molecules and targets in DOPIN; (ii) dock against pre-processed targets of the PDB; and (iii) search for off-targets. Maintained by the laboratory of Samy Meroueh at the Center for Computational Biology and Bioinformatics at the Indiana University School of Medicine.
- GPCRautomodel. Web service that automates the homology modeling of mammalian olfactory receptors (ORs) based on the six three-dimensional (3D) structures of G protein-coupled receptors (GPCRs) available so far and (ii) performs the docking of odorants on these models, using the concept of colony energy to score the complexes. Provided by INRA.
- kinDOCK. Allows comparative docking of ligands into the ATP-binding site of a protein kinase (target). A sequence alignment of the target and a protein kinase profile is performed using HMMER. It uses protein-protein superposition (automatically restricted to the ligand binding pocket) of the target three-dimensional structure with those of known complexes of protein kinases/ligands.
- iScreen. Web service for docking and screening the small molecular database on traditional Chinese medicine (TCM) on user's protein. iScreen is also implemented with the de novo evolution function for the selected TCM compounds using the LEA3D genetic algorithm
- idTarget. Web server for identifying biomolecular targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Maintained by the National Taiwan University.
- MetaDock. Online docking solution and docking results analysis service. Docking is done with GNU/GPL-licensed AutoDock v.4 and Dock6 under academic license
- Score. Allows to calculate some different docking scores of ligand-receptor complex that can be submitted as a whole file containing both interaction partners or as two separated files. The calculation phase is provided by VEGA. Provided by the Drug Design Laboratory of the University of Milano.
- Pose & Rank. Web server for scoring protein-ligand complexes. Provided by the laboratory of Andrej Sali.
- PLATINUM. Calculates hydrophobic properties of molecules and their match or mismatch in receptor–ligand complexes. These properties may help to analyze results of molecular docking.
Screening
Web services
- Blaster. Public access service for structure-based ligand discovery. Uses DOCK as the docking program and various ZINC Database subsets as the database.Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- AnchorQuery. Specialized pharmacophore search for targeting protein-protein interactions. Interactively search more than 20 million readily synthesizable compounds all of which contain an analog of a specific amino acid. Provided by the University of Pittsburgh.
- istar. Free web platform for large-scale protein-ligand docking based on the idock software. The web site can be downloaded and installed independently from GitHub. Developed by the Chinese university of Hong Kong.
- istar. Free web platform for large-scale protein-ligand docking based on the idock software. This link corresponds to the web site code that can be installed independently. Developed by the Chinese university of Hong Kong.
- GFscore. GFscore is a ranked-based consensus scoring function based on the five scoring functions : FlexX Score, G_Score, D_Score, ChemScore, and PMF Score available in TRIPOS Cscore module. The aim is to eliminate as many molecules as possible from proprietary in house database after a Virtual Library Screening (VLS) using TRIPOS FlexX for docking and the TRIPOS Cscore module for scoring. Developped and maintained by the Institute for Structural Biology and Microbiology, Marseille, France.
- Aggregator Advisor. Free web service to suggest molecules that aggregate or may aggregate under biochemical assay conditions. The approach is based on the chemical similarity to known aggregators, and physical properties. Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
Ligand design
Web services
- iScreen. Web service for docking and screening the small molecular database on traditional Chinese medicine (TCM) on user's protein. iScreen is also implemented with the de novo evolution function for the selected TCM compounds using the LEA3D genetic algorithm
- 3DLigandSite. Automated method for the prediction of ligand binding sites. Provided by the Imperial London College.
- PASS. Program for tentative identification of drug interaction pockets from protein structure.
- DEPTH. Web server to compute depth and predict small-molecule binding cavities in proteins
- VAMMPIRE-LORD. LORD (Lead Optimization by Rational Design) is a prediction tool based on the VAMMPIRE database (of matched molecular pairs) and using a atom-pair descriptor to represent the substitution environment. It operates on the principle that molecular transformations cause similar effects in similar substitution environments and is therefore able to extrapolate the knowledge of a given substitution effect to any similar system. LORD was implemented as an easy-to-use web server that guides the user step-by-step through the optimization process of a defined lead compound.