Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
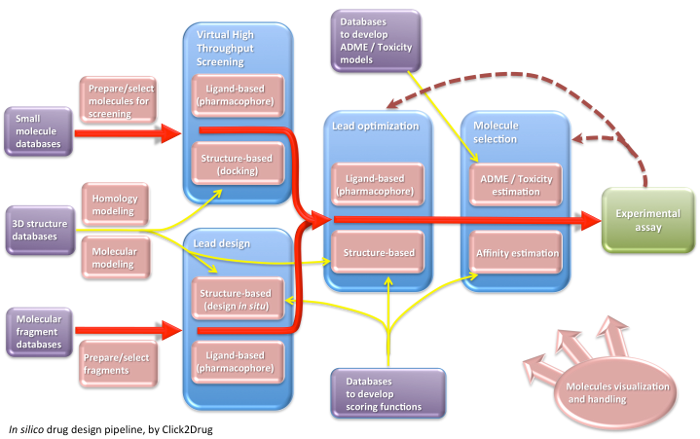
Show all links Hide all links
Homology Modeling
Software
- Modeller. Software for producing homology models of protein tertiary structures, using a technique inspired by nuclear magnetic resonance known as satisfaction of spatial restraints. Maintained by Andrej Sali at the University of California, San Francisco. Free for academic use. Graphical user interfaces and commercial versions are distributed by Accelrys.
- I-TASSER. Internet service for protein structure and function predictions. Models are built based on multiple-threading alignments by LOMETS and iterative TASSER simulations. I-TASSER (as 'Zhang-Server') was ranked as the No 1 server in recent CASP7 and CASP8 experiments. Exists as a standalone package. Provided by the University of Michigan.
- LOMETS. (Local Meta-Threading-Server). On-line web service for protein structure prediction. It generates 3D models by collecting high-scoring target-to-template alignments from 8 locally-installed threading programs (FUGUE, HHsearch, MUSTER, PPA, PROSPECT2, SAM-T02, SPARKS, SP3). Exists as a standalone package. Provided by the University of Michigan.
- MPACK. (Modeling Package). Integrated protein modeling suite that currently handles comparative and ab initio modeling procedures. The objective of this suite is to systematically bring different steps (or programs) under one roof in order to facilitate rapid model generation with minimal user effort and to create a biological data-flow pipeline for large scale-scale modeling of protein sequences from genomic projects. Provided by the University of Texas.
- ProModel. Allows homology modeling from either a selected template or a user defined template. Modeling in manual mode allows mutation, excision, deletion, insertion of residues or insertion of a loop by selecting the start and end anchors. Automated homology modeling can be performed by reading in the template file obtained from a local BLAST. ProModel enables analysis of the target protein structure, active site and channels. Provided by VLife.
- SCRWL. Program for prediction of protein sice chains prediction, based on the Dunbrack backbone-dependent rotamer library. Provided by the Dunbrack Lab.
- Biskit. Free and open source modular, object-oriented Python library for structural bioinformatics research that wraps external programs (BLAST, T-Coffee and Modeller) into an automated workflow. Developed by the institut Pasteur.
- ModPipe. Completely automated software pipeline that can calculate protein structure models for a large number of sequences with almost no manual intervention. In the simplest case, it takes as input a sequence identifier and a configuration file and produces one or more comparative models for that sequence. Free and open source software. Maintained by Andrej Sali at the University of California, San Francisco.
- RaptorX. Protein structure prediction program developed by Xu group, with a particular focus on the alignment of distantly-related proteins with sparse sequence profile and that of a single target to multiple templates. Currently, RaptorX consists of four major modules: single-template threading, alignment quality assessment, multiple-template threading and fragment-free approach to free modeling. Also exists as a web service.
- Prime. Fully-integrated protein structure prediction program, providing graphical interface, sequence alignment, secondary structure prediction, homology modeling, protein refinement, loop-prediction, and side-chain prediction. Developed by Schrödinger.
- ProSide. Predicts protein sidechain conformation. Since the residue-substitution by the target amino-acid sequence is possible, ProSide can be used also for simple homology modeling, in case there are neither insertion nor deletion. Can perform global optimization calculation of a complex, by putting ligand to a binding site, and optimizing positions and conformations of ligand and amino-acid sidechains. Distributed by IMMD.
- CABS. Versatile reduced representation tool for molecular modeling, including: de novo folding of small proteins, comparative modeling (especially in cases of poor templates) and structure prediction based on sparse experimental data. Developed by the Warsaw University.
Web services and databases
- SWISS-MODEL. Fully automated protein structure homology-modeling server, accessible via the ExPASy web server, or from the program DeepView (Swiss Pdb-Viewer).
- SWISS-MODEL Repository. Database of annotated three-dimensional comparative protein structure models generated by the fully automated homology-modelling pipeline SWISS-MODEL.
- Robetta. Web server. Rosetta homology modeling and ab initio fragment assembly with Ginzu domain prediction.
- ModWeb. Server for Protein Structure Modeling based on the Modeller program. Maintained by Andrej Sali at the University of California, San Francisco.
- I-TASSER. Internet service for protein structure and function predictions. Models are built based on multiple-threading alignments by LOMETS and iterative TASSER simulations. I-TASSER (as 'Zhang-Server') was ranked as the No 1 server in recent CASP7 and CASP8 experiments. Exists as a standalone package. Provided by the University of Michigan.
- RaptorX web server. Protein structure prediction web server developed by Xu group, with a particular focus on the alignment of distantly-related proteins with sparse sequence profile and that of a single target to multiple templates. Currently, RaptorX consists of four major modules: single-template threading, alignment quality assessment, multiple-template threading and fragment-free approach to free modeling. Due to limited computational power, this server offers the first three modules for regular usage. Also exists as a standalone program.
- TIP database. The Target Informatics Platform (TIP) database contains more than 195,000 high resolution protein structures and homology models, with annotated small molecule binding sites, covering major drug target families including proteases, kinases, phosphatases, phosphodiesterases, nuclear receptors, and GPCRs. The TIP database automatically and self-consistently updates itself, possibly including proprietary sequence and structure data. Developed and maintained by Eidogen-Sertanty, Inc.
- iProtein. iPad application providing access to the Eidogen-Sertanty's Target Informatics Platform (TIP).
- ModBase. Database of three-dimensional protein models calculated by comparative modeling. The models are derived by ModPipe, an automated modeling pipeline relying on the programs PSI-BLAST and MODELLER. The database also includes fold assignments and alignments on which the models were based. MODBASE also contains information about putative ligand binding sites, SNP annotation, and protein-protein interactions.
- ModEval. Model evaluation server for protein structure models. Maintained by Andrej Sali at the University of California, San Francisco.
- ModLoop. Web server for automated modeling of loops in protein structures. The server relies on the loop modeling routine in MODELLER that predicts the loop conformations by satisfaction of spatial restraints, without relying on a database of known protein structures. Maintained by Andrej Sali at the University of California, San Francisco.
- Protinfo ABCM. The Protinfo web server consists of a series of discrete modules that make predictions of, and provide information about, protein folding, structure, function, interaction, evolution, and design by applying computational methodologies developed by the Samudrala Computational Biology Research Group.
- PMP. (Protein Model Portal). Gives access to various models computed by comparative modeling methods provided by different partner sites, and provides access to various interactive services for model building, and quality assessment. Provided by the Swiss Institute of BioInformatics and the University of Basel.
- HHpred. Web server for homology detection & structure prediction by HMM-HMM comparison.
- CPHmodels. Protein homology modeling server. The template recognition is based on profile-profile alignment guided by secondary structure and exposure predictions. Maintained by the Center for Biological Sequence Analysis, Denmmark.
- GeneSilico Metaserver. Gateway to various methods for protein structure prediction, including primary structure, seconday structure, transmembrane helices, disordered regions, disulfide bonds, nucleic acid binding residues in proteins and tertiary structure. Maintained by the Bujnicki laboratory in IIMCB, Warsaw, Poland.
- QUARK. Internet service for ab initio protein folding and protein structure prediction, which aims to construct the correct protein 3D model from amino acid sequence only. QUARK models are built from small fragments (1-20 residues long) by replica-exchange Monte Carlo simulation under the guide of an atomic-level knowledge-based force field. QUARK was ranked as the No 1 server in Free-modeling (FM) in CASP9. Since no global template information is used in QUARK simulation, the server is suitable for proteins which are considered without homologous templates. Provided by the University of Michigan.
- SuperLooper. SuperLooper provides an online interface for the automatic, quick and interactive search and placement of loops in proteins. Loop candidates are selected from a database (LIMP) comprising ~ 180.000 loops of membrane proteins or, alternatively, from (LIP) containing ~ 513.000.00 segments of water-soluble proteins with lengths up to 35 residues. In addition to several filtering criteria regarding structural and sequence features, the software allows for placing the loop within the predicted membrane-water interface. Provided by Charité Berlin, Structural Bioinformatics Group.
- PEP-FOLD. De novo approach aimed at predicting peptide structures from amino acid sequences. This method, based on structural alphabet SA letters to describe the conformations of four consecutive residues, couples the predicted series of SA letters to a greedy algorithm and a coarse-grained force field. Developed by the University of Paris Diderot.
- FoldX. A whole package for protein modeling and design. Originally focused on the impact of mutation on protein structure stability based on an original force field strongly linked to mutagenesis data. It was developed by at the European Molecular Biology Laboratory in Heidelberg and at Center for Genomic Regulation in Barcelona. The FoldX Suite is available through academic or commercial license.
- LOMETS. (Local Meta-Threading-Server). On-line web service for protein structure prediction. It generates 3D models by collecting high-scoring target-to-template alignments from 8 locally-installed threading programs (FUGUE, HHsearch, MUSTER, PPA, PROSPECT2, SAM-T02, SPARKS, SP3). Exists as a standalone package. Provided by the University of Michigan.
- ESyPred3D. Automated homology modeling web server in which lignments are obtained by combining, weighting and screening the results of several multiple alignment programs. The final three dimensional structure is built using the modeling package MODELLER.
- MolProbity. Web service for all-atom structure validation for macromolecular crystallography. Maintained by the Richardson Lab, Duke University.
- PSiFR. (Protein Structure and Function predicton Resource) provides integrated tools for protein tertiary structure prediction and structure and sequence-based function annotation. The web portal provides access to TASSER, TASSER-Lite and MetaTASSER and DBD-Hunter, and the enzyme function inference engine EFICAz2.
- 3D-Jigsaw. Automated system to build three-dimensional models for proteins based on homologues of known structure.
- Geno3D. Automatic modeling of proteins three-dimensional structure using comparative protein structure modelling by spatial restraints (distances and dihedral) satisfaction. Provided by the Pole Bioinformatique Lyonnais.
- VADAR. (Volume, Area, Dihedral Angle Reporter) is a compilation of more than 15 different algorithms and programs for analyzing and assessing peptide and protein structures from their PDB coordinate data to quantitatively and qualitatively assess protein structures determined by X-ray crystallography, NMR spectroscopy, 3D-threading or homology modelling. Provided by the University of Alberta, Canada.
- phyre. (Protein Homology/analogY Recognition Engine). Automated 3D model building using profile-profile matching and secondary structure. Provided by the Structural Bioinformatics group, Imperial College London.
- HMMSTR/Rosetta. Predicts the structure of proteins from the sequence : secondary, local, supersecondary, and tertiary. Provided by the Depts of Biology & Computer Science, Rensselaer Polytechnic Institute
- GPCRautomodel. Web service that automates the homology modeling of mammalian olfactory receptors (ORs) based on the six three-dimensional (3D) structures of G protein-coupled receptors (GPCRs) available so far and (ii) performs the docking of odorants on these models, using the concept of colony energy to score the complexes. Provided by INRA.
- FALC-Loop. Web server for protein loop modeling using a fragment assembly and analytical loop closure method.
- IntFOLD. Web resource for protein fold recognition, 3D model quality assessment, intrinsic disorder prediction, domain prediction and ligand binding site prediction.
- HOMODELLER. Web server to predict protein 3D structure (PDB coordinates) from its primary sequence file by homology modelling. Provided by the University of Alberta, Canada.
- PEPstr. Web server to predict the tertiary structure of small peptides with sequence length varying between 7 to 25 residues. The prediction strategy is based on the realization that β-turn is an important and consistent feature of small peptides in addition to regular structures. Provided by the Bioinformatics Centre, Institute of Microbial Technology, Chandigarh.