Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
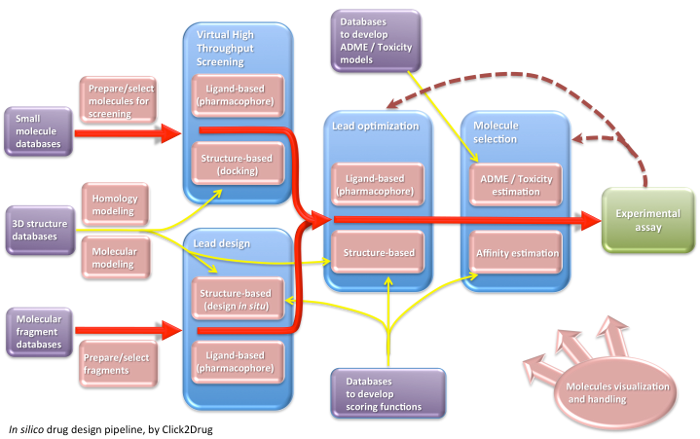
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
Show all links Hide all links
Databases
Databases handling
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Files can contains large amount of molecules and ChemDiff was test on files with up to 1 million ones. Free and open-source. Distributed by GGA software.
Chemical structure representations
File format Converters
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Files can contains large amount of molecules and ChemDiff was test on files with up to 1 million ones. Free and open-source. Distributed by GGA software.
- OSRA. (Optical Structure Recognition Application). Utility designed to convert graphical representations of chemical structures, as they appear in journal articles, patent documents, textbooks, trade magazines etc. OSRA can read a document in any of the over 90 graphical formats parseable by ImageMagick - including GIF, JPEG, PNG, TIFF, PDF, PS etc., and generate the SMILES or SDF representation of the molecular structure images encountered within that document. Free and open source. Developed by the Frederick National Laboratory for Cancer Research, NIH.
Screening
Software
- Pharmer. Free open source pharmacophore search technology that can search millions of chemical structures in seconds.
- Catalyst. Pharmacophore Modeling and Analysis; 3D database building and searching; Ligand conformer generation and analysis tools; Geometric, descriptor-based querying; Shape-based screening. Distributed by Accelrys as part of Discovery Studio.
- PharmaGist. Freely available web server for pharmacophore detection. The download version includes virtual screening capability.
- LiSiCA. LiSiCA (Ligand Similarity using Clique Algorithm) is a ligand-based virtual screening software that searches for 2D and 3D similarities between a reference compound and a database of target compounds which should be represented in a Mol2 format. The similarities are expressed using the Tanimoto coefficients and the target compounds are ranked accordingly. A PyMol plu-in is freely available, too. Developed by the University of Ljubljana, Slovenia.
- LigandScout. Fully integrated platform for virtual screening based on 3D chemical feature pharmacophore models. Developed by inte:ligand.
- CHAAC. Chaac is a ligand-based virtual screening tool. It compares your molecule with a database of ligands, and outputs a list of candidates with similar chemical profile to that of your query. Developed by Mind the Byte.
- IK. This virtual screening tool allows to compare in 3D molecules according to their behaviour with their environment. It generates a list of compounds similar to your query as output including also the non-structural analogues. Developed by Mind the Byte.
- KIZIN. Kizin supports compound selection. Given an input protein present in the ChEMBL database, and an internal or external library of drug candidates, it performs a 2D virtual screening, selecting molecules in the library likely to exhibit activity for that protein. Developed by Mind the Byte.
- ACPC. (AutoCorrelation of Partial Charges) Open source tool for ligand-based virtual screening using autocorrelation of partial charges. ACPC uses a rotation-translation invariant molecular descriptor.
- ChemCom. a computer application which facilitates searching and comparing chemical libraries. ChemCom aims to expedite the current, time consuming processes of comparing large, chemical databases. As such, this application can be used to speedup many processes including drug research and discovery. A free java web application is also available. Developed by the University of Kansas, USA.
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
- PyRX. Virtual Screening software for Computational Drug Discovery that can be used to screen libraries of compounds against potential drug targets. PyRx includes docking wizard with easy-to-use user interface which makes it a valuable tool for Computer-Aided Drug Design. PyRx also includes chemical spreadsheet-like functionality and visualization engine that are essential for Rational Drug Design. AutoDock 4 and AutoDock Vina are used as a docking software. Free and open source. For Windows, Linux and Mac OSX.
- MolSign. Program for pharmacophore identification and modeling. Can be used for querying databases as a pharmacophore based search. Provided by VLife.
- Spectrophores. Converts three-dimensional molecular property data (electrostatic potentials, molecular shape, lipophilicity, hardness and softness potentials) into one-dimensional spectra independent of the position and orientation of the molecule. It can be used to search for similar molecules and screen databases of small molecules. Open source software developed by Silicos.
- Shape-it. free open source shape-based alignment tool by representing molecules as a set of atomic Gaussians. Open source software developed by Silicos.
- Align-it. (Formerly Pharao). Pharmacophore-based tool to align small molecules. The tool is based on the concept of modeling pharmacophoric features by Gaussian 3D volumes instead of the more common point or sphere representations. The smooth nature of these continuous functions has a beneficent effect on the optimisation problem introduced during alignment. Open source software developed by Silicos.
- Open3DALIGN. Command-line tool aimed at unsupervised molecular alignment. Alignments are computed in an atom-based fashion (by means of a novel algorithm inspired to the LAMDA algorithm by Richmond and co-workers), in a pharmacophore-based fashion using Pharao as the alignment engine, or finally using a combination of the latter two methods. Free open source software. For Windows, Linux and Mac.
- Molegro Virtual Docker. The built-in Docking Template tool makes it possible to perform ligand-based screening by flexibly aligning a number of ligands (and determine a score for their similarity) and to perform hybrid docking by guiding the docking simulation by combining the template similarity score with a receptor-based docking scoring function. Distributed by Qiagen.
- GMA (Graph based Molecular Alignment). Combined 2D/3D approach for the fast superposition of flexible chemical structures. Part of the Chil2 suite. Open for general research.
- Fuzzee. Allows the identification of functionally similar molecules, based upon functional and structural groups or fragments. Part of the Chil2 suite. Open for general research.
- REDUCE. (Formerly FILTER). Tool to filter compounds from libraries using descriptors and functional groups. Part of the Molecular FORECASTER package, from Molecular Forecaster Inc.
- SELECT. (Selection and Extraction of Libraries Employing Clustering Techniques). Creates subset of libraries by diversity or similarity using clustering techniques. Part of the Molecular FORECASTER package.
- AutoclickChem. Computer program capable of performing click-chemistry reactions in silico. AutoClickChem can be used to produce large combinatorial libraries of compounds for use in virtual screens. As the compounds of these libraries are constructed according to the reactions of click chemistry, they can be easily synthesized for subsequent testing in biochemical assays. Exists as a web server. Distributed by the National Biomedical Computation Resource.
- REACTOR. (Rapid Enumeration by Automated Combinatorial Tool and Organic Reactions). Creates library of molecules by combining fragment libraries from a defined reaction, or from a generic attachment point on the fragments. Part of the Molecular FORECASTER package.
- FLAP. (Fingerprints for Ligands and Proteins). Provides a common reference framework for comparing molecules, using GRID Molecular Interaction Fields (MIFs). The fingerprints are characterised by quadruplets of pharmacophoric features and can be used for ligand-ligand, ligand-receptor, and receptor-receptor comparison. In addition, the quadruplets can be used to align molecules, and a more detailed comparison of the GRID MIF overlap calculated. When the template is a ligand, this enables ligand-based virtual screening and alignment. When the template is a receptor site, this enables structure-based screening and pose prediction. Provided by Molecular Discovery.
- GASP. Genetic Algorithm Similarity Program. Generates pharmacophores using a genetic algorithm. Distributed by Tripos.
- Tuplets. Pharmacophore-based virtual screening. Distributed by Tripos.
- KeyRecep. Estimates the characteristics of the binding site of the target protein by superposing multiple active compounds in 3D space so that the physicochemical properties of the compounds match maximally with each other. Can be used to estimate activities and vHTS. Distributed by IMMD.
- LigPrep. 2D to 3D structure conversions, including tautomeric, stereochemical, and ionization variations, as well as energy minimization and flexible filters to generate ligand libraries that are optimized for further computational analyses. Distributed by Schrodinger.
Web services
- SwissSimilarity. Web tool for rapid ligand-based virtual screening of small to unprecedented ultralarge libraries of small molecules. Screenable compounds include drugs, bioactive and commercial molecules, as well as 205 million of virtual compounds readily synthesizable from commercially available synthetic reagents. Predictions can be carried out on-the-fly using six different screening approaches, including 2D molecular fingerprints as well as superpositional and fast nonsuperpositional 3D similarity methodologies. SwissSimilarity is part of a large initiative of the SIB Swiss Institute of Bioinformatics to provide online tools for computer-aided drug design, such as SwissDock, SwissBioisostere or SwissTargetPrediction with which it can interoperate, and is linked to other well-established online tools and databases. User interface and backend have been designed for simplicity and ease of use, to provide proficient virtual screening capabilities to specialists and nonexperts in the field. The SwissSimilarity website, developed by the Molecular Modeling Group of SIB Swiss Institute of Bioinformatics, is accessible free of charge or login.
- Blaster. Public access service for structure-based ligand discovery. Uses DOCK as the docking program and various ZINC Database subsets as the database.Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- Aggregator Advisor. Free web service to suggest molecules that aggregate or may aggregate under biochemical assay conditions. The approach is based on the chemical similarity to known aggregators, and physical properties. Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- ZincPharmer. Free online interactive pharmacophore search software for screening the ZINC database. ZINCPharmer can import LigandScout and MOE pharmacophore definitions as well as perform structure-based pharmacophore elucidation.
- PUMA. Free web services that help at visulizing chemical space by computing molecular properties of pharmaceutical relevance, such as Murcko scaffolds, and performing diversity analysis. Developed and provided by the Department of Pharmacy of Universidad Nacional Autónoma de Mexico, Mexico.
- SimDOCK. Allows rapid selection of ligands fitting the active site of the submitted protein by superposition of its three-dimensional structure with those of known complexes of protein/ligands of the family.
- pep:MMs:MIMIC. Web-oriented tool that, given a peptide three-dimensional structure, is able to automate a multiconformers three-dimensional similarity search among 17 million of conformers calculated from 3.9 million of commercially available chemicals collected in the MMsINC database.
- AURAmol. Web service taking a candidate 2D or 3D molecular shape and use it to search for similarly shaped molecules in large databases. Provided by the University of York.
- SiMMap. Web server statistically deriving site-moiety map with several anchors, based on the target structure and several docked compounds. Each anchor includes three elements: a binding pocket with conserved interacting residues, the moiety composition of query compounds and pocket-moiety interaction type (electrostatic, hydrogen bonding or van der Waals). Compound highly agreeing with anchors of site-moiety map are expected to activate or inhibit the target protein.
- ShaEP. Free program to superimpose two rigid 3D molecular structure models, based on shape and electrostatic potentials, and computes a similarity index for the overlay. It can be used for the virtual screening of libraries of chemical structures against a known active molecule, or as a preparative step for 3D QSAR methods.
- AutoclickChem. Web server to perform click-chemistry reactions in silico. AutoClickChem can be used to produce large combinatorial libraries of compounds for use in virtual screens. As the compounds of these libraries are constructed according to the reactions of click chemistry, they can be easily synthesized for subsequent testing in biochemical assays. Exists as a stand alone program. Maintained by the National Biomedical Computation Resource.
Ligand design
Software
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
ADME Toxicity
Software
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
Web services
- PharmMapper. Freely accessed web-server designed to identify potential target candidates for the given probe small molecules (drugs, natural products, or other newly discovered compounds with binding targets unidentified) using pharmacophore mapping approach.