Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
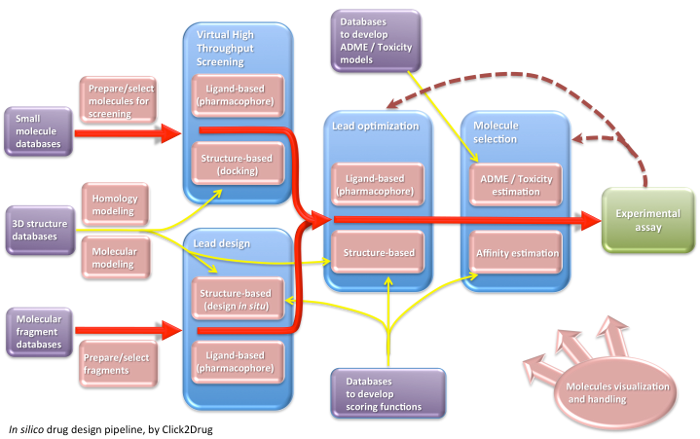
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
Show all links Hide all links
Molecular Modeling
Software
- CHARMM. (Chemistry at HARvard Macromolecular Mechanics). Package of molecular simulation programs, including source code and demos.
- GROMACS. (GROningen MAchine for Chemical Simulations). Free open source molecular dynamics simulation package.
- Amber. (Assisted Model Building with Energy Refinement). Package of molecular simulation programs, including source code and demos.
- AmberTools. AmberTools consists of several independently developed packages that work well by themselves, and with Amber itself. It contains NAB (build molecules; run MD or distance geometry, using generalized Born, Poisson-Boltzmann or 3D-RISM implicit solvent models), antechamber and MCPB (create force fields for general organic molecules and metal centers), tleap and sleap (Basic preparation program for Amber simulations), sqm (semiempirical and DFTB quantum chemistry program), pbsa (Performs numerical solutions to Poisson-Boltzmann models), 3D-RISM (Solves integral equation models for solvation), ptraj and cpptraj (structure and dynamics analysis of trajectories), MMPBSA.py and amberlite (Energy-based analyses of MD trajectories)
- OpenMM. High performance toolkit for CPU/GPU-accelerated molecular dynamics simulation and setup. Can be used as a library or as an application with an intuitive Python interface. Developed, maintained and provided open-source by Standford University, United States.
- VFFDT. It consists in a user-friendly Visual Force Field Derivation Toolkit (VFFDT) to derive the force field parameters via simply clicking on the bond or angle in the 3D viewer, and we have further extended our previous program to support the Hessian matrix output from a variety of quantum mechanics (QM) packages, including Gaussian 03/09, ORCA 3.0, QChem, GAMESS-US, and MOPAC 2009/2012. In this toolkit, a universal VFFDT XYZ file format containing the raw Hessian matrix is available for all of the QM packages, and an instant force field parametrization protocol based on a semiempirical quantum mechanics (SQM) method is introduced. The new function that can automatically obtain the relevant parameters for zinc, copper, iron, etc., which can be exported in AMBER Frcmod format, has been added. Furthermore, our VFFDT program can read and write files in AMBER Prepc, AMBER Frcmod, and AMBER Mol2 format and can also be used to customize, view, copy, and paste the force field parameters in the context of the 3D viewer, which provides utilities complementary to ANTECHAMBER, MCPB, and MCPB.py in the AmberTools.Proposed by the School of Pharmaceutical Sciences, Wenzhou Medical University
- NAMD. (NAnoscale Molecular Dynamics). Free parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. Based on Charm++ parallel objects.
- aMD. (accelerated Molecular Dynamics). enhanced-sampling method that improves the conformational space sampling by reducing energy barriers separating different states of a system. Distributed by the National Biomedical Computation Resource.
- MATCH. (Multipurpose Atom-Typer for CHARMM). Toolset of program libraries collectively titled multipurpose atom-typer for CHARMM (MATCH) for the automated assignment of atom types and force field paramters for molecular mechanics simulation of organic molecules. Developed by the Brooks lab, Michigan university.
- Desmond. Package to perform high-speed molecular dynamics simulations of biological systems on conventional computer clusters. Developed at D. E. Shaw Research.
- LAMMPS. Program for molecular dynamics. LAMMPS has potentials for soft materials (biomolecules, polymers), solid-state materials (metals, semiconductors) and coarse-grained or mesoscopic systems.
- MOLARIS-XG. MOLARIS-XG incorporates the methodologies of the former packages ENZYMIX and POLARIS. ENZYMIX is a macromolecular simulation program designed to study the functions of proteins ranging from ligand binding to free energy profiles of enzymatic reactions using the Empirical Valence Bond (EVB) approach and the Free Energy Perturbation (FEP) method. POLARIS is a fast converging computational software based on the Protein Dipoles-Langevin Dipoles (PDLD) approach, it is used for the calculation of free energies and electrostactic properties of molecules and macromolecules in solution through the evaluation of the permanent dipoles, induce dipoles, charges, dispersion contributions and hydrophobicity. Provided by the Warshel's group at the university of Southern California.
- OPENMD. Open source molecular dynamics to simulate liquids, proteins, nanoparticles, interfaces, and other complex systems using atom types with orientational degrees of freedom (e.g. sticky atoms, point dipoles, and coarse-grained assemblies).
- ORAC. Free open source program for Moleuclar Dynamics simulations. Maintained by the Florence university, Italy.
- AMMP VE. (Another Molecular Mechanics Program). Full-featured molecular mechanics, dynamics and modelling program that can manipulate both small molecules and macromolecules including proteins, nucleic acids and other polymers. Uses the VEGA ZZ interface. For MS Windows and Linux. Provided by the Drug Design Laboratory of the University of Milano.
- ACEMD (Accelerating bio-molecular simulations). Production bio-molecular dynamics (MD) software running on graphics processing units (GPUs) on NVIDIA graphics cards. ACEMD reads CHARMM/NAMD and AMBER input files. Distributed by Acellera. Free for 1 year for academic.
- CNS (Crystallography & NMR System). Provides a flexible multi-level hierachical approach for the most commonly used algorithms in macromolecular structure determination.
- Adun molecular simulation. Free biomolecular simulator developed at the Computational Biophysics and Biochemistry Laboratory, a part of the Research Unit on Biomedical Informatics of the UPF. It is distributed under the GNU General Public License.
- Tinker. Free, complete and general package for molecular mechanics and dynamics, with some special features for biopolymers.
- Force Field Explorer. Graphical user interface to the TINKER suite of molecular modeling tools.
- CHARMm. Commercial version of CHARMM with multiple graphical front ends.
- MacroModel. Commercial program for molecular modeling. Distributed by Schrodinger.
- MOIL. Public Domain Molecular Modeling Software, including energy calculations, energy minimization, molecular dynamics. Comes with a visualization program (zmoil) for graphic display of individual structures, dynamics, reaction paths and overlay of multiple structures, read PDB CRD DCD and (MOIL specific) PTH formatted files. For Windows, Mac and Linux.
- APBS. Adaptive Poisson-Boltzmann Solver (APBS) is a software for evaluating the electrostatic properties of nanoscale biomolecular systems.
- iAPBS. C/C++/Fortran interface to APBS. This interface enables access to most of APBS capability from within any C/C++ or Fortran code. In addition to the reference implementation, iAPBS/CHARMM, iAPBS/NAMD and iAPBS/Amber modules are also available. These modules extend CHARMM, NAMD and Amber functionality with APBS routines for electrostatic calculations. Provided by the McCammon Group, UCSD.
- Chemsol. Program to calculate solvation energies by using Langevin Dipoles (LD) model of the solvent and ab initio calculations. Also exists as a web service. Provided by the Warshel's group at the university of Southern California.
- BiKi. The BiKi Life Sciences suite involves several tools (e.g. accelerated binding/unbinding methods) for performing and analyzing MD specifically dedicated to medicinal chemists with the aim of simplifying drug discovery. Provided by BiKi Technologies, Italy.
- Abalone. General purpose molecular modeling program focused on molecular dynamics of biopolymers and molecular graphics. In addition, it can interact with external quantum chemical programs (NWChem, CP2K and PC GAMESS/Firefly. Provided by Agile Molecule.
- Ascalaph. General purpose molecular modeling suite that performs quantum mechanics calculations for initial molecular model development, molecular mechanics and dynamics simulations in the gas or in condensed phase. It can interact with external molecular modeling packages (MDynaMix, NWChem, CP2K and PC GAMESS/Firefly). Provided by Agile Molecule.
- HyperChem. Provides computational methods including molecular mechanics, molecular dynamics, and semi-empirical and ab-initio molecular orbital methods, as well as density functional theory.
- iHyperChem. Limited version of Professional HyperChem. This Level 1 version of iHyperChem allows creating and manipulating molecular systems and explore their structure. It also allows WiFi access to any Professional HyperChem server so that molecules, computations, and results can be transferred between the mobile client (iPhone or iPad) and the server. Provided by HyperChem.
- iHyperChem Free Version. Free Version of iHyperChem for iPad. Provided by HyperChem.
- Spartan. Provides computational methods including molecular mechanics, quantum mechanics, properties calculations (LogP, ovality, etc...), quantification of structural alignment using structure, chemical funtion descriptors or pharmacophore model, etc... Developed by Wavefunction, Inc.
- iSpartan. iSpartan is a versatile app for molecular modeling on the iPad, iPhone, and iPod Touch. Molecules are created by two-dimensional sketching and converted into a three-dimensional structure. Low-energy conformations can then be calculated and their geometries be queried. A database of 5,000 molecules (a subset of the Spartan Spectra and Properties Database, SSPD) can furthermore be accessed to obtain NMR and IR spectra, molecular orbitals, electrostatic potential maps, and other atomic and molecular properties. The database can be searched for substructures. Developed by Wavefunction, Inc.
- SCIGRESS. Desktop/server molecular modeling software suite that can apply a wide range of computational models to all types of molecular systems, from small organic molecules, to whole proteins, including linear scaling semiempirical quantum methods for protein optimization and ligand docking. Developed and distributed by Fujitsu, Ltd.
- TopoTools. TopoTools is a VMD plugin for manipulating topology information. It is meant to be a complementary tool to psfgen, which is very much optimized for building topologies for biomolecules. It makes access to the topology related data stored in VMD easily. It also has a number of high-level tools that allow reading and writing of topology file formats that cannot be parsed by the molfile plugins, parsing of parameter and residue database files for generation of complete input files for MD codes like LAMMPS and HOOMD-blue, and replicating or combining multiple systems. Developed by the Temple University, Philadelphia, USA.
- YASARA Dynamics. Adds support for molecular simulations to YASARA View/Model, Using the NOVA, YAMBER or AMBER force fields like AMBER. Provided by YASARA.
- Build model. Tool for creating protein models and their preparation for docking. Refine raw protein structure, add missing sidechains, assign protonation states of side chains at given pH, add missing hydrogen atoms, reconstruct crystallographically-related protein subunits and extract a reference ligand from the structure. Distributed by Moltech. For Windows and linux.
- Pdbfil. Automatically processes the protein coordinate data obtained from PDB for molecular calculations. Adds missing atoms, deletes unnecessary hetero-residues and water molecules, adds and optimizes hydrogen atoms. Atomic attributions like atomic charge or molecular force-field type are also automatically. Distributed by IMMD.
- Protein Preparation Wizard. Tool for correcting common structural problems and creating reliable, all-atom protein models. Distributed by Schrodinger.
- BALL. Biochemical Algorithms Library. Application framework in C++ designed for rapid software prototyping in the field of Computational Molecular Biology and Molecular Modeling. It provides an extensive set of data structures as well as classes for Molecular Mechanics, advanced solvation methods, comparison and analysis of protein structures, file import/export, and visualization. Free and opensource.
- pDynamo. pDynamo is an open source program library that has been designed for the simulation of molecular systems using quantum chemical (QC), molecular mechanical (MM) and hybrid QC/MM potential energy functions. Developed by the Institut de Biologie Structurale, Grenoble, France.
- Pcetk. Pcetk (a pDynamo-based continuum electrostatic toolkit) is a Python module extending the pDynamo library with a Poisson-Boltzmann continuum electrostatic model that allows for protonation state calculations in proteins. The module links pDynamo to the external solver of the Poisson-Boltzmann equation, extended-MEAD, which is used for the calculation of electrostatic energy terms. The calculation of protonation states and titration curves is done by using the module's own analytic or Monte Carlo routines or through an interface to the external sampling program, GMCT. Developed by the Institut de Biologie Structurale, Grenoble, France.
- MMTSB. Multiscale Modeling Tools for Structural Biology. Provides a collection of perl scripts for Structure preparation, Structure analysis, All-Atom Modeling, SICHO Lattice Modeling, Replica Exchange Sampling, Ensemble Computing and Structure Prediction.
- Computer-Aided Drug-Design Platform using PyMOL. PyMOL plugins providing a graphical user interface incorporating individual academic packages designed for protein preparation (AMBER package and Reduce), molecular mechanics applications (AMBER package), and docking and scoring (AutoDock Vina and SLIDE).
- pymacs. Python module for dealing with structure files and trajectory data from the GROMACS molecular dynamics package. It has interfaces to some gromacs functions and uses gromacs routines for command line parsing, reading and writing of structure files (pdb,gro,...) and for reading trajectory data (only xtc at the moment).
- PyRosetta. Interactive Python-based interface to the Rosetta molecular modeling suite. It enables users to design their own custom molecular modeling algorithms using Rosetta sampling methods and energy functions.
Web Services
- SwissParam. Provides topology and parameters for small organic molecules compatible with the CHARMM all atoms force field, for use with CHARMM and GROMACS.
- CHARMM-GUI. Provides (optimal, reasonable) CHARMM input files in a GUI fashion so that people can run the input on their machine, Helps people read and modify the input with their purposes, and provides also educational materials such as MM/MD lectures, as well as molecular animations. Provided by the University of Kansas.
- CHARMMing.org. CHARMMing contains an integrated set of tools for uploading structures, performing simulations, and viewing the results.
- LigParGen. Web-based service that provides OPLS-AA force field parameters for organic molecules or ligands. Beside PGR files, other ouput format includes parameters and topologies to be used with CHARMM, Gromacs, LAMMPS, CNS/X-PLOR, Q, DESMOND, BOSS, OpenMM and MCPRO. Molecules can be input in SMILES, MOL or PDB format with a maximum of 200 atoms. Provided also as a standalone program by the Department of Chemistry, University of Yale, United-States.
- LipidBuilder. A web-server based on a VMD plug-in and CHARMM force field to create, store and share lipid libraries. LipidBuilder automatically generates the topology and template of a given lipid. The lipid topology is created by combining the selected head group, extracted from a built-in library of structures and the provided hydrocarbon chains. Four different classes of hydrocarbons have been parametrized in the CHARMM force field: saturated, unsaturated, branched and cyclopropane. Developed by the EPFL, Lausanne, Switzerland.
- ParamChem. Provides topology and parameters for small organic molecules from CGenFF, for use with CHARMM and GROMACS. Provided by the University of Kentucky.
- PrimaDORAC. A free Web Interface for the Assignment of partial charges, chemical topology, and bonded parameters in small molecules to be used in molecular mechanics or molecular dynamics calculations. Provided by the University of Florence, Italy.
- MATCH server. The MATCH web server allows one to submit a molecule to generate both the topology and parameter file for a given small molecule. Molecule file formats: pdb, sdf, mol, mol2 are all accepted. Provided by the Brooks laboratory, University of Michigan.
- ProteinPrepare. a web application designed to interactively support the preparation of protein structures. Users can upload a PDB file, choose the solvent pH value, and inspect the resulting protonated residues and hydrogen-bonding network within a 3D web interface. Provided by Acellera, Ltd.
- MemBuilder. Prepares the initial configuration of a membrane model composed of different phospholipid molecules. This server is also dedicated to determine the lipid composition of each monolayer to support the asymmetry of the membrane bilayer. Provided by the Tarbiat Modares University.
- CABS-flex. Server for fast simulation of protein structure fluctuations. CABS-flex is a procedure for the simulation of structure flexibility of folded globular proteins. Using an input protein structure the CABS-flex outputs a set of protein models (reflecting the flexibility of the input structure, in all-atom PDB format) ready to use in structure-based studies of protein functions and interactions. Developed by the university of Warsaw.
- Dundee PRODRG2 Server. Converts coordinates for small molecules in PDB format to the following topology formats: GROMOS, GROMACS, WHAT IF, REFMAC5, CNS, O, SHELX, HEX and MOL2.
- PDB2PQR Server. Web server to convert PDB files into PQR files and perform an APBS calculation. Protein pKa are calculated using PROPKA.
- PROPKA. Web server to calculate pKa of protein titratable functions.
- PropKa. Web server to calculate pKa of protein titratable functions using PROPKA. Provided by the Drug Design Laboratory of the University of Milano.
- H++. Web server to compute pK values of ionizable groups in macromolecules and adds missing hydrogen atoms according to the specified pH of the environment.
- PDB_Hydro. Provides tools for mutating (change side-chains of a PDB file automatically, repair missing side-chains in a PDB file, construct polar hydrogen atoms and assig partial charges for electrostatic calculations) and solvating PDB files.
- POLYVIEW-MM. Web-based platform for animation and analysis of molecular simulations. Enables animation of trajectories generated by molecular dynamics and related simulation techniques, as well as visualization of alternative conformers, e.g. obtained as a result of protein structure prediction methods or small molecule docking.
- CLICK. Web server for superimposing the 3D structures of any pair of biomolecules (proteins, DNA, RNA, etc.). The server makes use of the Cartesian coordinates of the molecules with the option of using other structural features such as secondary structure, solvent accessible surface area and residue depth to guide the alignment. Help establishing protein relationships by detecting similarities in structural subdomains, domains and topological variants or to recognize conformational changes that may have occurred in structural domains or subdomains in one structure with respect to the other.
- SLITHER. Web server for generating contiguous conformations of substrate molecules entering into deep active sites of proteins or migrating across membrane transporters. Predicts whether a substrate molecule can crawl through an inner channel or a half-channel of proteins across surmountable energy barriers.
- AlloDeco. Server that implements a novel model for allostery. It computes the thermodynamic coupling between functional sites in proteins and then determines the contribution of specific interactions to that coupling. First, a 3-dimensional protein structure (PDB file) is transformed into a Gaussian Network Model. The coupled motions of two sites are then decomposed using Canonical Correlation Analysis. Finally, the statistical mechanics-based thermodynamic coupling function formalism is applied to identify interactions that mediate the thermodynamic coupling between the canonical pairs of motions. Developed at Weill Cornell Medical College, New York, United States.
- R.E.D. Server. Web service designed to automatically derive RESP and ESP charges, and to build force field libraries for new molecules/molecular fragments.
- Chemsol web service. Web service for the calculations of solvation free energies using the Langevin Dipoles (LD) solvation model, in which the solvent is approximated by polarizable dipoles fixed on a cubic grid.Also exists as a standalone program. Provided by the Warshel's group at the university of Southern California.
- MovieMaker. Web server that allows short (~10 sec), downloadable movies to be generated of protein dynamics. It accepts PDB files or PDB accession numbers as input and automatically outputs colorful animations covering a wide range of protein motions and other dynamic processes (simple rotation, morphing between two end conformers, short-scale, picosecond vibrations, ligand docking, protein oligomerization, mid-scale nanosecond (ensemble) motions and protein folding/unfolding). Note: MovieMaker is not a molecular dynamics server and does not perform MD calculations. Provided by the University of Alberta, Canada.
- Superpose. Protein superposition server, using a modified quaternion approach. From a superposition of two or more structures, it generates sequence alignments, structure alignments, PDB coordinates, RMSD statistics, Difference Distance Plots, and interactive images of the superimposed structures. Provided by the University of Alberta, Canada.
- ArbAlign. Web implmentation of the Kuhn-Munkres algorithm to optimally align two arbitrarily ordered isomers. Code and web interface provided by the Bucknell University, Lewisburg, USA.