Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
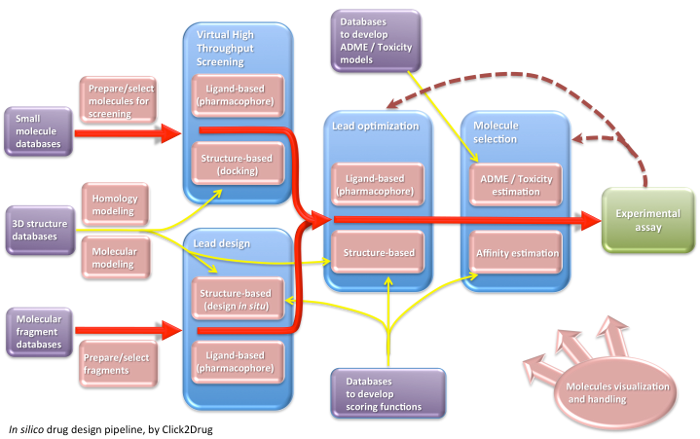
Show all links Hide all links
Databases
Databases handling
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Files can contains large amount of molecules and ChemDiff was test on files with up to 1 million ones. Free and open-source. Distributed by GGA software.
Chemical structure representations
2D drawing
- ChemDraw. Molecule editor developed by the cheminformatics company CambridgeSoft. For Windows and Mac.
- MarvinSketch. Advanced chemical editor for drawing chemical structures, queries and reactions developed by ChemAxon. Exists as an applet.
- ACD/ChemSketch. Molecule editor developed by ACD/Labs. Also available as freeware, with tools for 2D structure cleaning, 3D optimization and viewing, InChI generation and conversion, drawing of polymers, organometallics, and Markush structures. For Windows only.
- VLifeBase. Provides features to build a molecule from scratch using 2D Draw and conversion to 3D. The 3D editor allows addition, modification, replacement and deletion of atoms, bonds and groups, with Undo and Redo operations. Provided by VLife.
- ISIS/Draw. Chemical structure drawing program for Windows, published by MDL Information Systems. Free of charge for academic and personal use.
- JME Molecular Editor. Java applet which allows to draw / edit molecules and reactions (including generation of substructure queries) and to depict molecules directly within an HTML page. Editor can generate Daylight SMILES or MDL Molfile of created structures.
- Bioclipse. Java-based, open source, visual platform for chemo- and bioinformatics based on the Eclipse Rich Client Platform (RCP).
- Indigo-depict. Command-line molecule and reaction rendering utility. Free and open source. Distibuted by GGA software.
2D drawing online
- Marvin molecule editor and viewer. Java based chemical editor for drawing chemical structures. Includes unlimited structure based predictions for a range of properties (pKa, logD, name<>structure, etc.). Provided by ChemAxon.
3D viewers
- UCSF Chimera. Open source, highly extensible program for interactive visualization and analysis of molecular structures and related data. Free of charge for academic, government, non-profit, and personal use. For Windows, Mac and Linux. Developed by the Resource for Biocomputing, Visualization, and Informatics, UCSF.
- RasMol. Program for molecular graphics visualisation.
Definitions and syntax of file formats
- Daylight SMILES. SMILES (Simplified Molecular Input Line Entry System) is a line notation (a typographical method using printable characters) for entering and representing molecules and reactions.
- InChI. (IUPAC International Chemical Identifier) is a string of characters capable of uniquely representing a chemical substance. It is derived from a structural representation of that substance in a way designed to be independent of the way that the structure was drawn (thus a single compound will always produce the same identifier). It provides a precise, robust, IUPAC approved tag for representing a chemical substance.
- Tripos Mol2. Complete description of the Mol2 file format (.mol2).
- PDB format. Complete description of the PDB file format (.pdb).
- SDF format. Complete description of the SDF file format (.sdf).
- SMARTS format. SMARTS Tutorial by Daylight.
- OpenSMILES. Community sponsored open-standards version of the SMILES language for chemistry. OpenSMILES is part of the Blue Obelisk community.
File format Converters
- OpenBabel. Free open source chemical expert system mainly used for converting chemical file formats. For Windows, Unix, and Mac OS.
- Corina. Generates 3D structures for small and medium sized, drug-like molecules. Distributed by Molecular Networks.
- Indigo. Universal organic chemistry toolkit, containing tools for end users, as well as a documented API for developers. Free and open-source, but also available on a commercial basis. Distributed by GGA software.
- Indigo-depict. Command-line molecule and reaction rendering utility. Free and open source. Distibuted by GGA software.
- Indigo-cano. Command-line canonical SMILES generator. Free and open source. Distibuted by GGA software.
- Indigo-deco. Command-line program for R-Group deconvolution. Free and open source. Distibuted by GGA software.
- OMEGA. (Conformer Ensembles Containing Bioactive Conformations). Converts from 1D or 2D to 3D using distance bounds methods, with a focus on reproducing the bioactive conformation. Developed by OpenEye.
- COSMOS. (COordinates of Small MOleculeS). High-throughput method to predict the 3D structure of small molecules from their 1D/2D representations. Also exists as a web service. Provided by the University of california, Irvine.
- TorsionAnalyzer. Generate and analyse 3D conformers of small molecules. TorsionAnalyzer is based on an expert-derived collection of SMARTS patterns and rules (assigned peaks and tolerances). Rules result from statistical analysis of histograms derived from small molecule X-ray data extracted from the CSD. Rotatable bonds of molecules loaded into the TorsionAnalyzer are color-coded on the fly by means of a traffic light highlighting regular, borderline and unusual torsion angles. This allows the user to see at a glance if one or more torsion angles are out of the ordinary. Provided by BioSolveIT.
- LigPrep. 2D to 3D structure conversions, including tautomeric, stereochemical, and ionization variations, as well as energy minimization and flexible filters to generate ligand libraries that are optimized for further computational analyses. Distributed by Schrodinger.
- CACTVS. Universal scriptable toolkit for chemical information processing. Used by PubChem. Maintained and distributed by Xemistry. Free for academic.
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Files can contains large amount of molecules and ChemDiff was test on files with up to 1 million ones. Free and open-source. Distributed by GGA software.
- OSRA. (Optical Structure Recognition Application). Utility designed to convert graphical representations of chemical structures, as they appear in journal articles, patent documents, textbooks, trade magazines etc. OSRA can read a document in any of the over 90 graphical formats parseable by ImageMagick - including GIF, JPEG, PNG, TIFF, PDF, PS etc., and generate the SMILES or SDF representation of the molecular structure images encountered within that document. Free and open source. Developed by the Frederick National Laboratory for Cancer Research, NIH.
- MayaChemTools. Collection of Perl scripts, modules, and classes to support day-to-day computational chemistry needs. Free software, open source. Provided by Manish Sud.
- VLife Engine. Engine module of VLifeMDS containing basic molecular modeling capabilities such as building, viewing, editing, modifying, and optimizing small and arge molecules. Fast conformer generation by systematic and Monte-carlo methods. Provided by VLife.
- SPORES. (Structure PrOtonation and REcognition System). Structure recognition tool for automated protein and ligand preparation. SPORES generates connectivity, hybridisation, atom and bond types from the coordinates of the molecule`s heavy atoms and hydrogen atoms to the structure. The protonation can either be done by just adding missing hydrogen atoms or as a complete reprotonation. SPORES is able to generate different protonation states, tautomers and stereoisomers for a given structure. Developed by the Konstanz university.
- DG-AMMOS. Program to generate 3D conformation of small molecules using Distance Geometry and Automated Molecular Mechanics Optimization for in silico Screening. Freely distributed by the University of Paris Diderot.
- Key3D. Molecular modeling tool to convert 2D structures (chemical structural formula) of compounds drawn by ISIS-Draw or ChemDraw to 3D structures with additional information on atomic charge etc. Distributed by IMMD.
- ChemDoodle. A software suite for drawing chemical structure diagrams, including the ability to calculate NMR spectra, generate IUPAC names and line notations for structures, manipulate structures imported from the Internet, interpret and interconvert files generated by other chemical drawing software programs, illustrate glassware and equipment setups, and draw TLC plates. Distributed by iChemLabs LLC.
- CONFLEX. Software for searching and analyzing the conformational space of small and large molecules.
- JOElib. Cheminformatics library mainly used for conversion of file formats. Written in Java. For Windows, Unix, and Mac OS.
- CDK (Chemistry Development Kit). LGPL-ed library for bio- and cheminformatics and computational chemistry written in Java. Opensource.
- MolEngine. .NET Cheminformatics Toolkit completely built on Microsoft .NET platform. By using Mono, MolEngine can run on other platform, such as Mac, Linux, iPad. Distributed by Scilligence.
- Indigo. Universal organic chemistry toolkit. Free and opensource. Provided by GGA.
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Provided by GGA.
- Open Drug Discovery Toolkit. ODDT is a free and open source tool for both computer aided drug discovery (CADD) developers and researchers. It reimplements many state-of-the-art methods, such as machine learning scoring functions (RF-Score and NNScore) and wraps other external software to ease the process of developing CADD pipelines. ODDT is an out-of-the-box solution designed to be easily customizable and extensible. Therefore, users are strongly encouraged to extend it and develop new methods. Provided by the Institute of Biochemistry and Biophysics PAS, Warsaw, Poland.
- RDKit. Collection of cheminformatics and machine-learning software written in C++ and Python.
- Mol2Mol. Molecule file manipulation and conversion program.
- Fconv. Molecule file manipulation and conversion program.
- Knodle. KNOwledge-Driven Ligand Extractor is a software library for the recognition of atomic types, their hybridization states and bond orders in the structures of small molecules. Its prediction model is based on nonlinear Support Vector Machines. The process of bond and atom properties perception is divided into several steps. At the beginning, only information about the coordinates and elements for each atom is available: (i) Connectivity is recognized; (ii) A search of rings is performed to find the Smallest Set of Smallest Rings (SSSR); (iii) Atomic hybridizations are predicted by the corresponding SVM model; (iv) Bond orders are predicted by the corresponding SVM model; (v) Aromatic cycles are found and (vi) Atomic types are set in obedience to the functional groups. Some bonds are reassigned during this stage. Linux and MacOS version are free of charge. Maintained by the Nano-D team, Inria/CNRS Grenoble, France.
- smi23d. Consists of two programs that can be used to convert one or more SMILES strings to 3D. For Mac and Linux. Also exists as a web service.
- Scaffold Hunter. JAVA-based software tool for exploring the chemical space by enabling generation of and navigation in a scaffold tree hierarchy annotated with various data. The graphical visualization of structural relationships allows to analyze large data sets, e.g., to correlate chemical structure and biochemical activity. Free open source software developed and supported by the Chair of algorithm Engineering at Technical University Dortmund and the Department of Chemical Biology at Max-Planck Institute for Molecular Physiology Dortmund.
- ScaffoldTreeGenerator. Java-based program which generates the scaffold tree database independently of Scaffold Hunter. Free open source software developed and supported by the Chair of algorithm Engineering at Technical University Dortmund and the Department of Chemical Biology at Max-Planck Institute for Molecular Physiology Dortmund.
- Strip-it. Program to extract scaffolds from organic drug-like molecules by 'stripping' away sidechains and representing the remaining structure in a condensed form. Open source software distributed by Silicos.
- fragmentizer. Free and open source python script that can decompose PDBs of small-molecule compounds into their constituent fragments. Developed by the National Biomedical Computation Resource.
- Epik. Enumerates ligand protonation states and tautomers in biological conditions. Distributed by Schrodinger.
- iBabel. iBabel is an alternative graphical interface to Open Babel for Macintosh OS X.
- PerlMol. Collection of perl modules providing objects and methods for representing molecules, atoms, and bonds in Perl; doing substructure matching; and reading and writing files in various formats.
- The SDF Toolkit in Perl 5. The purpose of this SDF toolkit is to provide functions to read and parse SDFs, filter, and add/remove properties.
Web services
- E-Babel. Online version of OpenbBabel. Maintained by the Virtual Computational Chemistry Laboratory.
- Corina online demo. Online demo of CORINA. Generates 3D coordinates from SMILES.
- Chemical Identifier Resolver. Converts a given structure identifier into another representation or structure identifier, using CACTVS. May give the name of a given molecule from SMILES of InChi, thanks to a database of 68 million chemical names linked to 16 million unique structure records.
File format Converters
- COSMOS. (COordinates of Small MOleculeS). High-throughput method to predict the 3D structure of small molecules from their 1D/2D representations. Also exists as a standalone program. Provided by the University of california, Irvine.
Web services
- VEGA WE. Web server for file translation tool, properties and surface calculation. Provided by the Drug Design Laboratory of the University of Milano.
- PDB Hydrogen Addition. Tool to add the hydrogen in a given PDB (for protein, DNA and drugs).
- DG-AMMOS. Generates single 3D conformer for small compound.
- Frog2. FRee Online druG conformation generation.
- Smiles2Monomers. Smiles2Monomers is a software to infer monomeric structure of polymers from their atomic structure. The web server is available for peptide-like compounds in the second tab and provides an interface to upload a compound in the SMILES format to compute the monomeric structure in two different formats: text formats (the structure is downloadable in json and xml) or image format (the colored picture of the monomeric structure mapped on the atomic structure is directly available in the browser or downloadable into a zip file). Provided by the University of Lille, France.
- e-LEA3D. Draw a molecule by using the ACD applet (v.1.30) and generate 3D coordinates by using the program Frog.
- MolEdit. Web server for 2D molecular editor & 3D structure optimization. Provided by the Drug Design Laboratory of the University of Milano.
- Chemozart. Chemozart is a 3D Molecule editor and visualizer built on top of native web components. It offers an easy to access service, user-friendly graphical interface and modular design. It is a client centric web application which communicates with the server via a representational state transfer style web service. Both client-side and server-side application are written in JavaScript. A combination of JavaScript and HTML is used to draw three-dimensional structures of molecules. Provided by the Department of Chemistry, Shahid Beheshti University, Tehran, Iran
- ProBuilder. Protein/peptide builder from 1D to 3D. Provided by the Drug Design Laboratory of the University of Milano.
- Online SMILES Translator and Structure File Generator. Translates SMILES into SDF, PDB of MOL formats, possibly generating 3D coordinates.
- smi23D web service. Translates SMILES strings or a URL to a SMILES file and get back the 3D coordinates in SDF. users can get the SDF file by typing directly the SMILES in the web browser, e.g. http://rest.rguha.net/threed/d3.py/get3d?smiles=c1ccccc1
- Smi2Depict. Webservice to generate 2D images from SMILES.
- GIF/PNG-Creator. GIF/PNG-Creator for 2D Plots of Chemical Structures from SMILES or structure files, using CACTVS. Maintained by the National Cancer institute, NIH.
- depict. Webservice using the molconvert tool of ChemAxon to generate 2D images from SMILES.
- SMARTSviewer. Webservice to visualize 2D images from SMARTS.
- OSRA web service. (Optical Structure Recognition Application). Web service designed to convert graphical representations of chemical structures, as they appear in journal articles, patent documents, textbooks, trade magazines etc. OSRA can read a document in any of the over 90 graphical formats parseable by ImageMagick - including GIF, JPEG, PNG, TIFF, PDF, PS etc., and generate the SMILES or SDF representation of the molecular structure images encountered within that document. Free and open source. Developed by the Frederick National Laboratory for Cancer Research, NIH.
Screening
Software
- CoLibri. Assembles huge compound collections from multiple sources and various input formats into a virtual screening library, removes duplicates, assesses the distribution of physico-chemical properties of the compounds and makes selections/filter based on any property-threshold, molecules name-pattern or presence/absence of a particular substructure motif. Generates fragments library. Modifies molecules or fragments for generating, transforming and general handling of virtual screening libraries. Distributed by BioSolveIT.
- Corina. Generates 3D structures for small and medium sized, drug-like molecules. Distributed by Molecular Networks.
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
- LigPrep. 2D to 3D structure conversions, including tautomeric, stereochemical, and ionization variations, as well as energy minimization and flexible filters to generate ligand libraries that are optimized for further computational analyses. Distributed by Schrodinger.
- Balloon. Free command-line program that creates 3D atomic coordinates from molecular connectivity via distance geometry and confomer ensembles using a multi-objective genetic algorithm. The input can be SMILES, SDF or MOL2 format. Output is SDF or MOL2.
- Epik. Enumerates ligand protonation states and tautomers in biological conditions. Distributed by Schrodinger.
Ligand design
Software
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
ADME Toxicity
Software
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.